Chapter 33: Amyloidosis
AMYLOIDOSIS
Definition:
The original amyloid name is derived from the waxy, starch-like property of the material.
Amyloidosis is a misnomer, as it was originally thought that the fibrils were starch like in nature.
Amyloidosis is the accumulation of various insoluble fibrillar proteins between the cells of tissues to the extent that affects the function.
Amyloidosis is a generic term that refers to diverse extracellular proteins, which are insoluble fibrillar proteins, arranging themselves as non-branching β-pleated sheets that are resistant to proteolysis and phagocytosis.
The accumulation of amyloid within tissues and organs leads to loss of function and organ enlargement.
Amyloid is a protein metabolism disorder in which there is the extracellular deposition of pathological insoluble fibrillar protein in organs and tissues.
Amyloid is β-pleated sheets that are responsible for its insolubility and resistance to proteolysis. These fibrils generally represent proteolytic fragments of various plasma proteins and are non-branching, and possess a β-pleated sheet structure.
This amyloid material is characterized by:
- Common morphologic properties.
- Affinity for specific dye-congo-red.
- Characteristic appearance under polarized light.
With Light Microscope and Haematoxylin/Eosine stain:
Amyloid appears as an amorphous, eosinophilic, hyaline, extracellular substance with progressive accumulation, encroaching on and producing pressure atrophies of the adjacent cells.
Amyloidosis
Physical nature of amyloid:
Amyloidosis is not a single disease. It is a group of diseases having in common the deposition of similar appearing proteins.
Amyloid with light microscopy and standard stains show an amorphous, eosinophilic, hyaline, extracellular substance with progressive atrophy of adjacent cells.
Congo red can differentiate amyloid material.
By E/M microscopy, amyloid is seen to be made up largely of non-branching fibrils of indefinite length and diameter of roughly 7.5 to 10 nm.
X-ray-crystallography and infrared spectroscopy show characteristic β-pleated sheets.
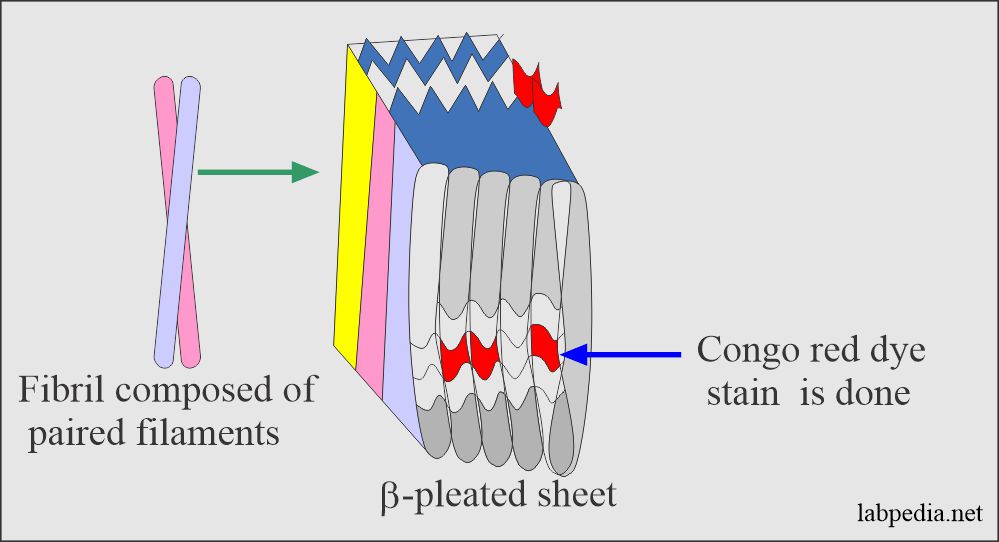
Fig 176: Physical Structure of Amyloid Material
Chemical Nature of amyloid:
- 95% of the amyloid material consists of fibril proteins, and 5% is the P-component, a glycoprotein.
- P component protein looks like C-reactive proteins. This is different from the amyloid fibrils.
- All fibrils have a p component associated with them, derived from the serum amyloid P component, a plasma protein closely related to C-reactive proteins.
- There are 15 biochemically distinct amyloid proteins are identified, and two of these are more common, and these are:
- Amyloid light chain (AL) is derived from the plasma cells.
- It consists of immunoglobulin light chains.
- It consists of only light chain immunoglobulins.
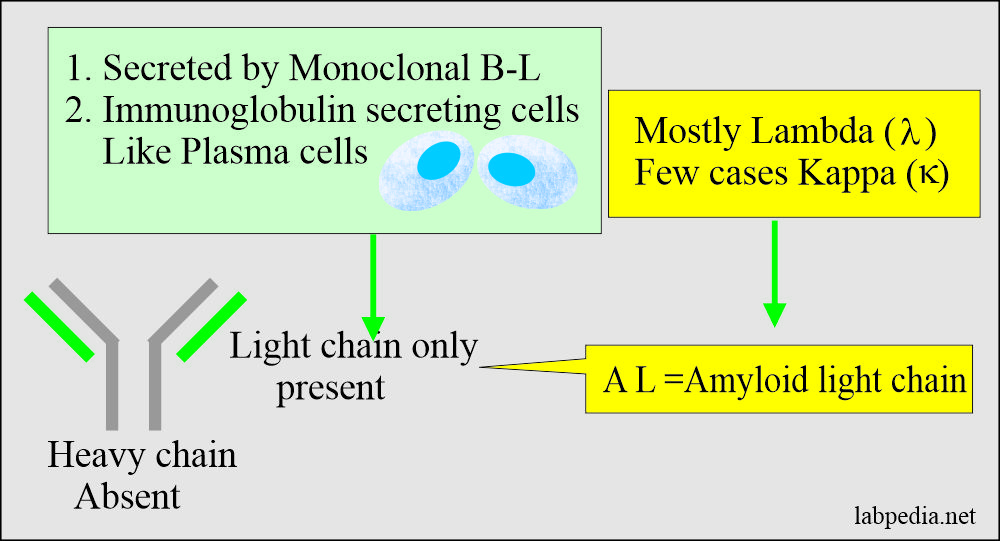
Amyloidosis: Amyloid light chain (AL) protein
- Amyloid-associated (AA) protein is a non-immunoglobulin protein synthesized in the liver.
- This is an amyloid fibrillary protein (AA), is different than immunoglobulins.
- It has a molecular weight of 8500 and consists of 76 amino acids residue.
- This is considered secondary amyloidosis.
- This is derived from the serum-amyloid-associated proteins synthesized in the liver, and it circulates in the blood with HDL3 subclass of lipoproteins.
- There are more amyloid proteins seen in amyloidosis, are:
- Transthyretin.
- β2-microglobulin.
- β2- Amyloid protein (Aβ).
- Deposition of amyloid occurs mainly in the following condtions:
- Primary amyloidosis: In patients with monoclonal B cell proliferations.
- Secondary amyloidosis: In patients with the chronic inflammatory process like rheumatoid arthritis.
- In hereditary circumstances like neuropathies.
- In patients being maintained on chronic dialysis.
- In patients with Alzheimer’s disease.
- Amyloid light chain (AL) is derived from the plasma cells.
Classification
There are 15 biochemically distinct forms, but the most common are the following.
- AL (Amyloid Light Chain).
- AA (Amyloid associated non-Ig-proteins which forms in the liver).
- AB-Found in Alzheimer’s disease.
Classification by Type of Proteins
- AL Amyloid (Amyloid light chain): This type consists of the usually variable region of the light chain of immunoglobulin. It is mostly derived from λ (Lambda) and rarely from K (Kappa) chains. This condition is associated with:-
- Multiple myeloma (10-15% of myeloma patients develop AL amyloid).
- B-cell lymphoma.
- AA Amyloid (Amyloid associated): It is derived from acute-phase proteins and is commonly found in patients with chronic inflammatory diseases, neoplastic diseases, and hereditary disorders, which leads to so-called secondary amyloidosis.
Clinical Classification
This is based on clinical presentation. These are of the following types.
- Primary Amyloidosis
This is the most common type of amyloidosis. It appears without any preceding disease. In 1/3 cases, it is due to plasma cell neoplasm. Also seen in B-cell lymphoma. Light chains of immunoglobulin molecules are excessively produced by neoplastic plasma cells either as an idiopathic disease or as part of Frank’s multiple myeloma.
- Secondary Amyloidosis
There is the deposition of serum protein (serum amyloid A or AA). It is due to previous chronic inflammatory diseases like rheumatoid arthritis, system lupus erythematosus, ankylosing spondylitis. Other chronic infectious diseases are:- Tuberculosis, lung abscess, and chronic osteomyelitis.
This may also be seen in Hodgkin’s Lymphoma and Renal cell carcinoma.
- Isolated or Localized Amyloidosis
It tends to involve a single organ or tissue. e.g., in Alzheimer’s disease, amyloid is only in the brain and its blood vessels. This is β-amyloid protein.
- In Down Syndrome, this may be seen in features like Alzheimer’s in all patients, but the age is 35 years.
- In Diabetes Mellitus, Amyloid deposits are seen in islet cells of Langerhan’s. It is derived from peptides related to a variant of calcitonin.
- In Senile Cardiac Amyloidosis, This may occur in men after the age of 70 years.
Pathogenesis
Amyloidosis pathogenesis may be simply by some stimuli which give rise to insoluble fibrils or unknown is due to the chronic inflammatory process.
In reactive systemic amyloidosis, it appears that long-standing tissue destruction and inflammation leads to elevated serum AA (amyloid associated) proteins.
Serum amyloid associated (AA) is synthesized in the liver under the influence of IL-1 and IL-6.
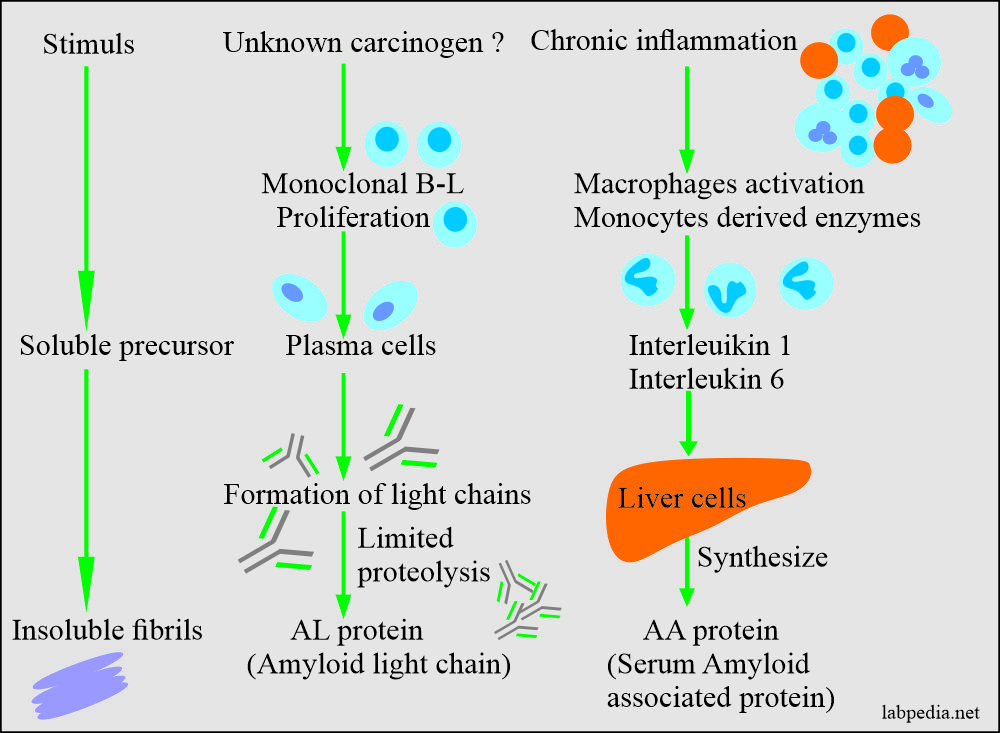
Fig 177: Pathogenesis of Amyloidosis
Clinical Features
- The S/S depends upon the involvement of the organs.
- Sometimes there are no S/S, and the patient may have a sudden death.
- There may be clinical manifestations, and amyloidosis ultimately leads to death.
- Non-specific S/S are—weakness, weight loss, light-headedness, or syncope.
- Specific S/S appears late, and these are related to the organ involved, e.g., Heart, GIT, or Renal system.
- There may be hepatomegaly and splenomegaly, and alteration in the serum protein level may be seen.
- Renal involvement is dominating and is a life-threatening feature in systemic amyloidosis.
- It gives rise to proteinuria and important cause of the nephrotic syndrome.
- Progressive obliteration of the glomeruli in advanced cases leads to uremia and renal failure.
- Cardiac amyloidosis is also common in the AL type and less common in the reactive systemic amyloidosis.
- This may be seen in the advanced age group.
- Its presentation is congestive heart failure.
- The main involvement is the conduction system and leads to arrhythmias.
- GIT involvement is insidious.
- When there is the tongue’s involvement, that will lead to enlargement and inelasticity, which will cause difficulty in speaking and swallowing.
- When the stomach and intestine are involved, that will lead to malabsorption, diarrhea, and indigestion.
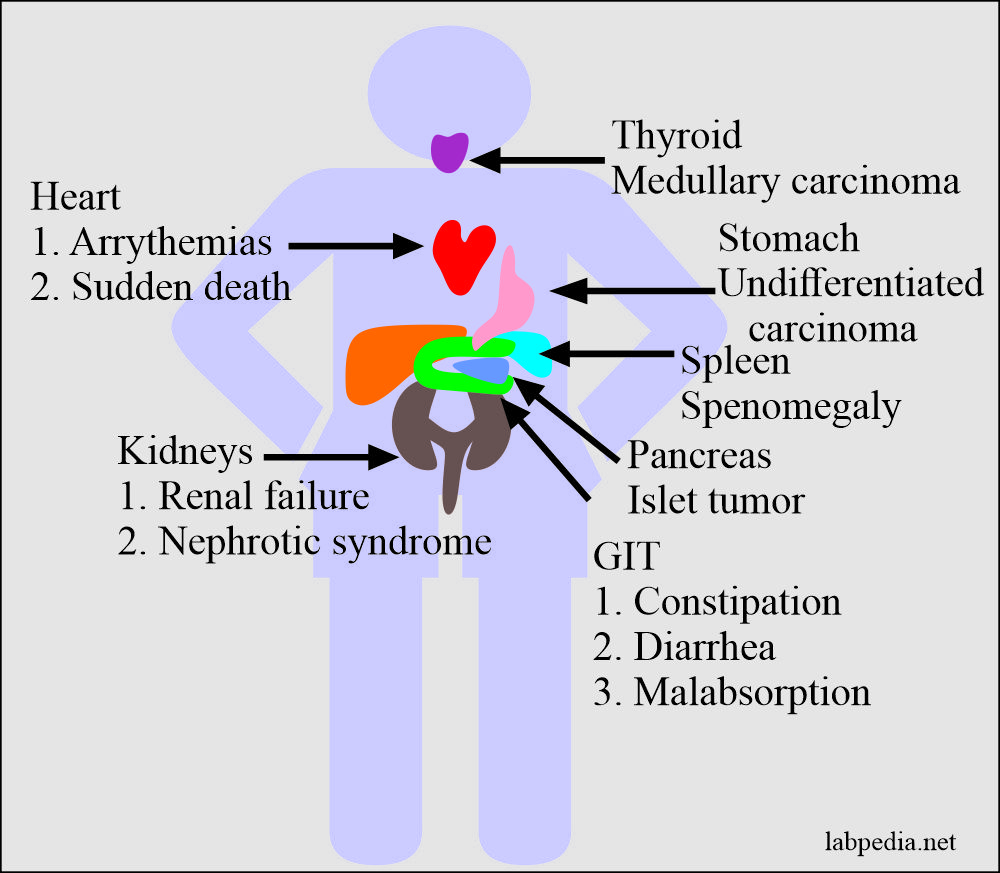
Amyloidosis signs and symptoms
Consequences of Amyloidosis
Amyloid fibrils, when first deposited, are usually in close association with the subendothelial basement. The amyloid deposit occurs between parenchymal cells and then around the blood vessels supply. Extensions of these deposits eventually entrap the parenchymal cells and interfere with their nutrition and strangulate those cells. Ultimately, atrophy takes place.
Commonly Affected Organs
Kidney
This may occur in-patient with multiple myeloma and chronic, long-standing inflammatory disorders. Clinically these patients develop nephrotic syndrome. Because of progressive glomerular obliteration, renal failure ultimately takes place.
Heart
This is usually seen in the systemic form of amyloidosis. There is entrapment of the conduction system, which leads to arrhythmias and sudden death.
Gastrointestinal Tract
Amyloidosis involves ganglia, smooth muscles, and vasculature. Clinically patients will complain of either constipation or diarrhea. Occasionally there may be signs of malabsorption.
Endocrines
The localized form of amyloidosis may be found in certain endocrines tumors like:-
- Medullary carcinoma of the thyroid.
- Islets tumor of the pancreas.
- Undifferentiated carcinoma of the stomach.
Diagnosis
- Rectal biopsy, which will be positive for congo-red stain. Other sites are gingival or kidneys.
- Up to 80% of primary amyloidosis patients may have low-level paraproteins in the serum or urine.
Treatment
- The reversal of the deposition of amyloid is difficult.
- Management should stop further deposition, which may be achieved by Melphalan and corticosteroids.
- In the case of secondary amyloidosis, the underlying cause may be treated.
- The nephrotic syndrome and cardiopulmonary failure need relevant therapies.
- If there is a source of infection that may be treated accordingly.
- Colchicine may help familial Mediterranian fever.
- Chemotherapy shows some benefits in AL amyloidosis.
Prognosis
In the systemic form of amyloidosis, the patient course is usually unremitting and ultimately fatal.
Patients with multiple myeloma and AL amyloidosis generally die within 1-2 years.
Patients with AA amyloidosis may die within 5 years due to cardiac or renal failure.